
Nucleotide Sequence Databases
The Data Format In Nucleotide Sequence Databases
Learning objectives:
- Understand the flatfile formats used in biological sequence databases.
Introduction
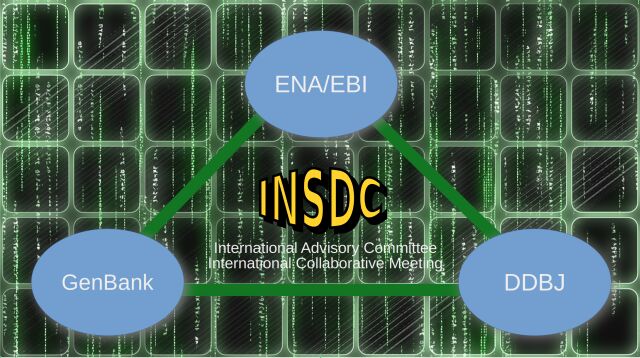
For bioinformaticians, understanding the management and exchange of nucleotide sequence data is crucial. The primary repositories for such data are the databases affiliated with the International Nucleotide Sequence Database Collaboration (INSDC): DDBJ, EMBL Now, European Nucleotide Archive (ENA) since 1993, and GenBank. These databases share daily data, ensuring that new or updated information is disseminated every 24 hours. This seamless exchange is facilitated by standard data formats, particularly the foundational text file known as a flatfile, a text file that orchestrates the transfer of information between these databases.
Flatfiles serve as the elementary format for information within sequence databases, allowing for straightforward data transfer between different entities. The consistency in flatfile formats enables a one-to-one mapping of fields from one format to another. Over time, various file formats have been employed, with some gaining widespread usage while others fading into obsolescence. The success of a format hinges on its adaptability across contexts and its effectiveness in encapsulating and representing diverse biological data, which is essential for archival and communication among scientists.
Every database entry comprises information about a specific sequence, encompassing details such as a sequence description, the originating organism, and relevant bibliographic references. The sequence can represent an individual gene, a gene segment, a whole genome shotgun (WGS), or various RNA forms (mRNA, tRNA, ncRNA, or rRNA) presented as cDNA. Additionally, the database may include sequences derived from environmental sources with unidentified organisms as their origin.
FASTA-Format Files
In its simplest form, a sequence record can be represented as a string of nucleotides accompanied by a primary tag or identifier. Among the commonly used formats is FASTA, a straightforward representation initially introduced as part of the FASTA software suite in 1985. The format is user-friendly for both human interpretation and computer processing. A bare FASTA sequence record appears as follows:
>NR_033657.1
CCCTTTGTGAGGCGCGGGAGAGATGTGCACCCCACATCTCTTTGCTTGAGTCCTGGTCCGCAAGATGGGT
GCTTGTTCGTGTGCCCCACCCCACCCCCACGCCCGAGGCCGGGTGCGGCAACTCCGCCCGCCAGCCGCTC
CTGCATTAAAATTTCATGGGCGCTGCAGTAAACGCGAAGACGACGCCGGTTGCGCTGGGGGGCCGTAGGG
AGGGAGGCTGCGGACCAGAGCGGGGAGGAGTTTGCGAGGGGAGGCGCTTGGGAGGACTCCCCCAGTTCTG
TTAAGAGCTGGGCACAAAGAGCCCTTTGCACTCCAGCCGCAGGAGCGAGGAGGAGCCGAGCTAGATGCAC
ATGGGCGTGGGGACTCCGGTACAACACCAACCCACAAGGTTAGCAGCCCTTACCATTCCTCCACTTATGG
Simple Definition Line
Here, the ">" character indicates the start of a new sequence record, and the unique identifier (NR_033657.1) is followed by the nucleotide sequence. The accession number (NR_033657.1) is a constant identifier associated with the sequence and should be cited in publications. The version number suffix lets users quickly discern the most up-to-date record, incrementing each time the sequence is updated.
In nucleotide sequence databases, such as GenBank, the ".1" in a sequence identifier like "NR_033657.1" refers to the version number of that particular sequence. The identifier is typically composed of two parts: the accession number (in this case, "NR_033657") and the version number (".1").
The accession number after the ">" character is a unique identifier assigned to a specific sequence submitted to the database. It remains constant and is associated with that particular sequence for its lifetime. Researchers cite accession numbers in publications and communications to refer to specific genetic sequences.
The version number is appended to the accession number and represents different versions or updates of the same underlying sequence. When a sequence undergoes updates or revisions, the version number is incremented. The example "NR_033657.1" indicates the sequence's first version with the accession number "NR_033657." If further modifications or annotations are made to the sequence, a new version would be assigned, such as "NR_033657.2," and so on.
The version numbering system allows users to track changes to a particular sequence over time. It permits preserving a clear and organized record of the sequence's history within the database.
Expanded Definition Line
For enhanced informativeness, the definition line can be expanded as follows:
>Genbank | NR_033657.1 Mus musculus myocardial infarction associated transcript
(non-protein coding) (Miat), transcript variant 1, long non-coding RNA
Additional details may include the source database (Genbank) and accession in this modified FASTA definition line. Version number (NR_033657.1) and a concise description of the biological entity represented by the sequence provides a richer understanding of the data.
Including the source database in a FASTA definition line is optional and depends on the level of detail or metadata provided by the submitter or curator of the sequence. While the example provided included information about the source database ("Genbank" in ">Genbank|Genbank | NR_033657.1 Mus musculus myocardial infarction associated transcript (non-protein coding) (Miat), transcript variant 1, long non-coding RNA"), it's not a mandatory component of a FASTA definition line.
In some cases, the submitter may provide only essential information, such as the unique identifier (accession. version number), a brief description of the sequence, and perhaps information about the organism or gene represented. Including the source database is a matter of preference. It can depend on the goals of the database, the nature of the data, and the conventions followed by the submitting institution.
However, when the source database information is available, it can be valuable for users as it provides context about where the sequence is sourced from and offers insights into curation and quality control processes specific to that database. Researchers often appreciate additional metadata that aids in interpreting and utilizing genetic information.
The Line Length
The line length after the ">" character is optional in a FASTA format but must be a single line. The ">" character indicates the beginning of a new sequence record, and what follows is typically the sequence data. The sequence can be represented as a continuous string of nucleotides, or line breaks can be introduced for better readability.
Traditionally, sequences are formatted with a specified line length, often around 60 to 80 characters per line. This line length convention helps with visual inspection and manual editing of the sequences. However, this formatting is optional, and sequences can be presented without line breaks.
For example, a sequence with line breaks might look like this:
>NR_033657.1
CCCTTTGTGAGGCGCGGGAGAGATGTGCACCCCACATCTCTTTGCTTGAGTCCTGGTCCGCAAGATGGGT
GCTTGTTCGTGTGCCCCACCCCACCCCCACGCCCGAGGCCGGGTGCGGCAACTCCGCCCGCCAGCCGCTC
CTGCATTAAAATTTCATGGGCGCTGCAGTAAACGCGAAGACGACGCCGGTTGCGCTGGGGGGCCGTAGGG
AGGGAGGCTGCGGACCAGAGCGGGGAGGAGTTTGCGAGGGGAGGCGCTTGGGAGGACTCCCCCAGTTCTG
TTAAGAGCTGGGCACAAAGAGCCCTTTGCACTCCAGCCGCAGGAGCGAGGAGGAGCCGAGCTAGATGCAC
ATGGGCGTGGGGACTCCGGTACAACACCAACCCACAAGGTTAGCAGCCCTTACCATTCCTCCACTTATGG
However, it is also valid to represent the sequence as a single continuous line, like this:
>NR_033657.1
CCCTTTGTGAGGCGCGGGAGAGATGTGCACCCCACATCTCTTTGCTTGAGTCCTG...
The decision on line length often depends on the preferences of the user or the specific requirements of the software or database being used to process the sequence data. Regardless of the line length, the primary focus is maintaining the correct sequence information and associated metadata in the FASTA format.
The FASTA format's simplicity, readability, versatility, compatibility, efficiency, historical significance, ease of implementation, and widespread adoption contribute to its popularity in bioinformatics. It remains a fundamental and widely accepted format for representing biological sequence information.
The Multi-FASTA File Format
The multi-FASTA file format is an extension of the standard FASTA format used in bioinformatics to store and organize multiple sequences in a single file. In a multi-FASTA file, each sequence is represented by a block of text following the standard FASTA conventions, including a header line starting with the ">" symbol, which contains a unique identifier and optional description, followed by the sequence data.
The key distinction of a multi-FASTA file is that it can contain more than one sequence. These sequences are typically separated by the ">" character at the beginning of a new header line, designating the start of a new sequence record. Each subsequent block of text represents a distinct sequence in the file. For example:
>Sequence_A
ATCGATCGATCG...
>Sequence_B
GCTAGCTAGCTA...
In this example, "Sequence_A" and "Sequence_B" are distinct sequences in the multi-FASTA file.
The multi-FASTA format is handy in bioinformatics analyses that simultaneously handle and process multiple sequences. It allows researchers to organize related sequences, such as those from different genes or species, in a single file, facilitating efficient data management and analysis.
Bioinformatics tools and software designed to work with FASTA files can often handle multi-FASTA files, making it a convenient and widely accepted format for various applications, including sequence alignment, homology searches, and phylogenetic analysis. The multi-FASTA format simplifies the organization and storage of sequence data, contributing to its popularity in the bioinformatics community.
The File Extensions Used
FASTA files typically use the ".fa" or ".fasta" file extensions. These extensions help users and software identify the file format and the type of data contained within. ".fa" and ".fasta" are commonly accepted, and the choice between them is often a matter of personal or institutional preference.
For example, a FASTA file named "sequences.fa" or "data.fasta" would be recognizable as a file containing biological sequence information in the FASTA format.
It's important to understand that while these are the standard conventions, the actual file extension does not affect the content or structure of the FASTA file. The format is determined by the content and adherence to the FASTA conventions, such as the use of ">" to indicate sequence headers and the arrangement of sequence data.
Nucleotide Sequence Flatfiles
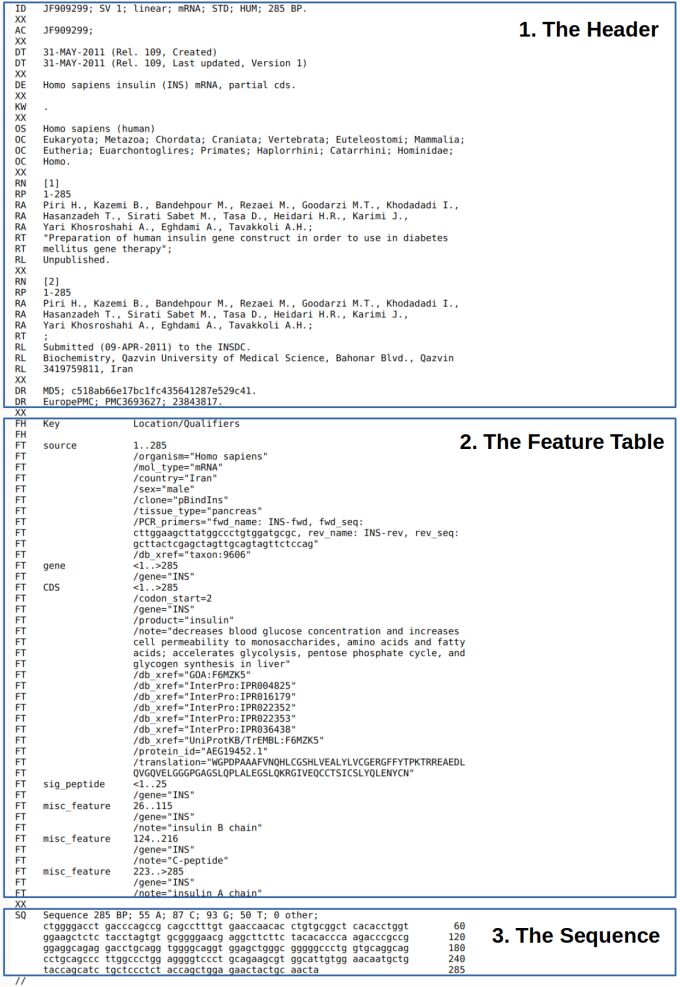
Nucleotide Sequence Flatfiles serve as the foundational information units in sequence databases, which are pivotal in facilitating seamless information exchange across these databases. An in-depth understanding of the composition of flatfiles is crucial to unraveling the wealth of information they contain. These flatfiles, while exhibiting minor format differences between databases, can be dissected into three significant components, each serving a distinct purpose and contributing to the comprehensive representation of genetic data.
The first and foremost component is the header, a critical section that encapsulates information and descriptors about the entire record. Within the header, researchers and database users can find essential metadata such as the origin of the sequence, taxonomic information, and any experimental details related to the data. It is a comprehensive introduction, offering context and background for the subsequent information embedded in the flatfile (Figure 1.).
The second component, the feature table, emerges as a critical element in enhancing the interpretability of the genetic sequence. This section provides detailed annotations that shed light on various features embedded within the sequence. Researchers can extract valuable insights into gene locations, functional elements, and any notable characteristics associated with the genetic material. The feature table acts as a navigational guide, allowing users to identify and comprehend specific elements within the sequence quickly (Figure 1.).
The third and final component is the sequence, representing the core genetic information encoded in the flatfile. This section contains the nucleotide sequence, allowing researchers to perform myriad analyses, ranging from sequence alignments to evolutionary studies. The sequence component is the heart of the flat file, where the raw genetic data is in a format that is readily utilizable for diverse research endeavors (Figure 1.).
The Header
We use the ENA-flatfile format as an example (Figure 1.). We have included DDBJ and Genbank flatfiles in the Appendix. The header part of the ENA file contains crucial metadata and descriptive information about the genetic sequence. It is a comprehensive introduction to the sequence data, providing details that help researchers understand the genetic information's origin, characteristics, and context.
The header is the most database-specific part because it contains information specific to the database's organization and data structure. The accession number, version information, creation dates, and other details are specific to the database system (such as the European Nucleotide Archive or ENA). Different databases may have distinct formats and requirements for these header fields. The header serves as a metadata framework designed by the database to catalog and organize the genetic sequences it hosts. It allows for efficient retrieval, interpretation, and integration of sequence data within the context of the specific database system.
The header includes various lines that contribute to this informative section:
ID Line:
ID JF909299; SV 1; linear; mRNA; STD; HUM; 285 BP.
The ID line contains the accession number (JF909299), version (SV 1), molecule type (mRNA), topology (linear), species code (HUM for Homo sapiens), and sequence length (285 base pairs). It functions as a unique identifier and provides fundamental information about the sequence. The version (SV 1) corresponds to the version notation JF909299.1. The Genbank and DDBJ use the name LOCUS line instead of the ID line in ENA format.
AC Line:
AC JF909299;
The AC line specifies the accession number (JF909299), a unique identifier assigned to the sequence. This accession number is used for referencing and retrieval of the sequence.
DT Lines:
DT 31-MAY-2011 (Rel. 109, Created)
DT 31-MAY-2011 (Rel. 109, Last updated, Version 1)
The DT lines include creation and update dates, as well as release information. These dates are crucial for tracking when the sequence was initially created, last updated, and the version released.
Note that the version on the DT line does not always correspond to the "accession.version" (JF909299.1). When changes only affect features without modifying the nucleotide sequence, the "accession.version" does not change. The "accession.version" is a unique identifier for a specific version of the sequence, and it typically increments only when there are changes to the underlying nucleotide sequence.
In scenarios where modifications involve annotations, metadata, or other non-sequence-related features, the "accession.version" remains constant, allowing for a clear distinction between different versions of the same sequence while reflecting the nature of the changes made. The version number is crucial for tracking the history and evolution of the genetic sequence in the database.
DE Line:
DE Homo sapiens insulin (INS) mRNA, partial cds.
The DE line provides a descriptive entry, offering information about the genetic content, including the organism (Homo sapiens), gene (INS), and the nature of the sequence (mRNA, partial cds).
The DDBJ/GenBank DEFINITION Line looks like this:
DEFINITION Homo sapiens insulin (INS) mRNA, partial cds.
While the specific syntax and position within the flatfile differ, these lines aim to offer a brief but informative description of the genetic sequence in a standardized manner across different nucleotide sequence databases. Researchers and users can expect similar information in the DE line of ENA and the DEFINITION line of DDBJ/GenBank.
While a substantial portion can be automatically generated from various sections within the record, a meticulous review process is implemented to guarantee the preservation of consistency and the richness of information. Encapsulating the entirety of the biological context associated with a sequence within a single line of text is a challenging task. However, a wealth of information will promptly unfold in subsequent sections of the same record
KW Line:
The KW line typically contains keywords describing the sequence's content. In this case, it is empty.
OS and OC Lines:
The OS line indicates the organism source (Homo sapiens), while the OC lines provide the organism classification hierarchy, specifying the taxonomic categories of the organism.
The taxonomic categories are listed hierarchically from broader classifications to more specific ones. The general order is as follows:
- Domain: The highest taxonomic rank, representing the broadest classification of life forms.
- Kingdom: A more specific classification under the domain.
- Phylum: Further refinement of the classification within a kingdom.
- Class: A more specific classification under a phylum.
- Order: A taxonomic rank under a class.
- Family: A taxonomic rank under an order.
- Genus: A taxonomic rank under a family.
- Species: The most specific classification, representing individual organisms within a genus.
Each level in the hierarchy narrows down the classification, providing a more specific designation for the organism. The OC lines use this hierarchical structure to convey the taxonomic relationships and classifications of the organism associated with the genetic sequence.
The Reference Blocks In The Header
Reference blocks refer to sections within the flatfile format that provide information about the references or sources associated with a particular genetic sequence. These references help users understand the context, origin, and relevant literature related to the sequence data. The reference information is typically structured, often with multiple reference blocks for different sources.
In an ENA flatfile, reference blocks include:
- Several lines offer details about the source of the sequence.
- The authors of the related research or study.
- The title of the work.
- Additional information, such as publication status and location.
The critical components of reference blocks may include:
- RN (Reference Number) Line:
- Indicates the reference number or identifier associated with the specific reference.
- RP (Reference Position) Line:
- Specifies the position or range of positions on the sequence corresponding to the referenced work.
- RA (Reference Author) Line:
- Lists the names of the authors who contributed to the referenced work.
- RT (Reference Title) Line:
- Provides the title of the referenced work, offering insights into the focus of the study.
- RL (Reference Location) Line:
- Includes information about the publication status, journal name, and other details about where the referenced work is available
Reference blocks enhance the utility and reliability of sequence databases by linking genetic information to the broader scientific literature, fostering transparency, and facilitating accurate interpretation and use of the data. Including references or citations in a sequence record depends on various factors, including the nature of the data, the sequence's source, and the specific database's practices.
While many sequence records do include references to the original literature, primarily when the sequence is associated with a specific study or publication, there are cases where sequences are submitted without explicit references. For example:
- User-submitted Data: TIn cases where users submit their sequences to a database, they might only sometimes provide detailed references.
- Automatically Generated Data: Some sequences are generated through automated processes, such as genome annotation pipelines, where the information might be derived from computational analyses without directly associating with a specific publication.
- Consensus or Derived Sequences: Databases may include consensus sequences or sequences derived from multiple sources, where the original contributors may not be explicitly cited.
While references can provide valuable context and attribution, their inclusion may vary based on the database policies and the nature of the data. However, in scientific research, it is generally encouraged to provide references when submitting sequences to databases, as it enhances transparency, allows for proper attribution, and facilitates the validation of data by other researchers. Database administrators often encourage users to include relevant literature information when submitting sequences.
The header is the most database-specific part because it contains information specific to the database's organization and data structure. The accession number, version information, creation dates, and other details are specific to the database system (such as the European Nucleotide Archive or ENA). Different databases may have distinct formats and requirements for these header fields. The header serves as a metadata framework designed by the database to catalog and organize the genetic sequences it hosts. It allows for efficient retrieval, interpretation, and integration of sequence data within the context of the specific database system.
RN and RP Lines:
RN [1]
RP 1-285
The RN and RP lines represent a reference entry number (RN) and the corresponding reference position (RP) on the sequence. These lines link the sequence to relevant references.
RA, RT, and RL Lines:
RA Piri H., Kazemi B., Bandehpour M., Rezaei M., Goodarzi M.T., Khodadadi I.,
...
RL Unpublished.
The RA line contains author information, the RT line provides the title of the reference, and the RL line offers details about the publication status and location of the reference.
DR and FH Lines:
DR MD5; c518ab66e17bc1fc435641287e529c41.
DR EuropePMC; PMC3693627; 23843817.
The DR lines cross-reference other databases, linking the sequence to related resources. The FH line introduces the Feature Table (FT) that follows.
The Feature Table
This section of the ENA feature table provides comprehensive information about the source of the nucleotide sequence.
This standardized representation, known as the feature table, comprises feature keys (single words or abbreviations indicating specific biological properties), location information indicating where the feature is situated within the sequence, and additional qualifiers offering descriptive details about the feature. The comprehensive online documentation for the INSDC feature table provides detailed information on permissible features and the qualifiers that can be associated with each feature. The language used in the feature table adheres to common biological research terminology, ensuring consistency across entries in DDBJ, ENA, and GenBank.
Let's break down the meaning and notation of each line:
...
FH Key Location/Qualifiers
FH
FT source 1..285
FT /organism="Homo sapiens"
FT /mol_type="mRNA"
FT /country="Iran"
FT /sex="male"
FT /clone="pBindIns"
FT /tissue_type="pancreas"
FT /PCR_primers="fwd_name: INS-fwd, fwd_seq:
FT cttggaagcttatggccctgtggatgcgc, rev_name: INS-rev, rev_seq:
FT gcttactcgagctagttgcagtagttctccag"
FT /db_xref="taxon:9606"
...
FH Key Location/Qualifiers:
- FH: Feature Header, indicating the beginning of the feature table section.
- Key Location/Qualifiers: Describing the subsequent lines as keys, locations, or qualifiers for features.
FT source 1..285:
- FT: Feature Table, indicating the start of a new feature.
- source: Describes the feature as the source of the sequence.
- 1..285: Specifies the location of the source feature, spanning from position 1 to 285 in the sequence.
FT /organism="Homo sapiens":
- Specifies a qualifier for the source feature.
- Indicates that the organism associated with the source is Homo sapiens (human).
FT /mol_type="mRNA":
- Specifies another qualifier for the source feature.
- Indicates that the molecule type is messenger RNA (mRNA).
FT /country="Iran":
- Provides additional information about the source.
- Specifies the country of origin as Iran.
FT /sex="male":
- Specifies the sex of the individual from whom the mRNA was derived.
- Indicates that the source is male.
FT /clone="pBindIns":
- Specifies the clone information for the source.
- Indicates that the sequence is derived from a clone labeled as "pBindIns."
FT /tissue_type="pancreas":
- Specifies the tissue type of the source.
- Indicates that the mRNA is derived from the pancreas.
FT /PCR_primers="fwd_name: INS-fwd, fwd_seq: ... rev_name: INS-rev, rev_seq: ...":
- Provides information about the PCR primers used in the sequencing process.
- Specifies the forward primer name, sequence, reverse primer name, and sequence.
FT /db_xref="taxon:9606":
- Specifies a cross-reference to a taxonomic database.
- Indicates that the organism's taxon ID is 9606, corresponding to Homo sapiens.
In summary, this feature table section describes the source of the sequence, including details such as organism, molecule type, country, sex, clone information, tissue type, PCR primers, and a cross-reference to the taxonomic database. The notation with slashes ("/") and key-value pairs provides a structured format for presenting this detailed information in the ENA feature table.
FT gene <1..>285
FT /gene="INS"
The "FT" indicates the start of a new feature, and the "gene" Describes the feature as a gene. "<1..>285" Specifies the location of the gene feature, indicating that it spans from position 1 to 285 in the sequence. The associated gene is identified as "INS," indicating that the nucleotide sequence corresponds to the insulin gene.
FT CDS <1..>285
FT /codon_start=2
FT /gene="INS"
FT /product="insulin"
FT /note="decreases blood glucose concentration and increases
FT cell permeability to monosaccharides, amino acids and fatty
FT acids; accelerates glycolysis, pentose phosphate cycle, and
FT glycogen synthesis in liver"
FT /db_xref="GOA:F6MZK5"
FT /db_xref="InterPro:IPR004825"
FT /db_xref="InterPro:IPR016179"
FT /db_xref="InterPro:IPR022352"
FT /db_xref="InterPro:IPR022353"
FT /db_xref="InterPro:IPR036438"
FT /db_xref="UniProtKB/TrEMBL:F6MZK5"
FT /protein_id="AEG19452.1"
FT /translation="WGPDPAAAFVNQHLCGSHLVEALYLVCGERGFFYTPKTRREAEDL
FT QVGQVELGGGPGAGSLQPLALEGSLQKRGIVEQCCTSICSLYQLENYCN"
This part of the ENA (European Nucleotide Archive) feature table provides comprehensive information about the coding sequence (CDS) associated with the nucleotide sequence, including its location, codon start, functional notes, cross-references to external databases, protein identifier, and the amino acid translation of the encoded protein. Let's break down the content:
FT CDS <1..>285:
- "CDS": Describes the feature as a coding sequence.
- "<1..>285": Specifies the location of the CDS feature, indicating that it spans from position 1 to 285 in the sequence.
FT /codon_start=2:
- Specifies a qualifier for the CDS feature.
- Indicates that the codon start is at position 2. This information is crucial for interpreting the reading frame and correctly translating the nucleotide sequence into amino acids.
FT /gene="INS":
- Specify another qualifier for the CDS feature.
- Indicates the gene associated with this coding sequence as "INS," which stands for insulin.
FT /product="insulin":
- Specifies the product of the CDS, which is the insulin protein.
FT /note="...":
- Provide additional notes about the CDS.
- Describes the functional aspects of the insulin protein, including its effects such as decreasing blood glucose concentration, increasing cell permeability to monosaccharides, amino acids, and fatty acids, as well as its roles in accelerating glycolysis, pentose phosphate cycle, and glycogen synthesis in the liver.
FT /db_xref="GOA:F6MZK5" ... /db_xref="UniProtKB/TrEMBL:F6MZK5":
- Specifies cross-references to external databases.
- Includes references to the Gene Ontology Annotation (GOA) database and UniProtKB/TrEMBL, providing additional resources for information about the protein.
FT /protein_id="AEG19452.1":
- Specifies the protein identifier associated with this CDS in the ENA database.
*FT /translation="WGPDPAAAFVNQHLCGSHLVEALYLVCGERGFFYTPKTRREAEDL ... QVGQVELGGGPGAGSLQPLALEGSLQKRGIVEQCCTSICSLYQLENYCN":
- Provides the amino acid translation of the CDS.
- Specifies the sequence of the translated insulin protein, which consists of the amino acids represented by the codons in the specified range.
It's important to note that while cross-references are generally accurate, users should exercise caution and critically evaluate the information based on their specific research needs. The accuracy of cross-references is also subject to the dynamic nature of scientific knowledge, and updates may occur as new information emerges. You should consult the original databases and literature for the most up-to-date and accurate information.
FT sig_peptide <1..25
FT /gene="INS"
FT misc_feature 26..115
FT /gene="INS"
FT /note="insulin B chain"
FT misc_feature 124..216
FT /gene="INS"
FT /note="C-peptide"
FT misc_feature 223..>285
FT /gene="INS"
FT /note="insulin A chain"
This section of the ENA feature table provides detailed information about various features associated with the insulin gene, including a signal peptide, the insulin B chain, the C-peptide, and the insulin A chain. Let's break down the content:
FT sig_peptide <1..25:
- "FT": Feature Table, indicating the start of a new feature.
- "sig_peptide": Describes the feature as a signal peptide.
- "<1..25": Specifies the location of the signal peptide, indicating that it spans from position 1 to 25 in the sequence.
FT /gene="INS":
- Specifies a qualifier for the signal peptide feature.
- Indicates the gene associated with this signal peptide as "INS," which is the insulin gene.
FT misc_feature 26..115:
- "misc_feature": Describes a miscellaneous feature.
- "26..115": Specifies the location of the miscellaneous feature, indicating that it spans from position 26 to 115 in the sequence.
FT /gene="INS" /note="insulin B chain":
- Specifies qualifiers for the miscellaneous feature.
- Indicates that the gene associated with this feature is "INS," representing the insulin B chain.
FT misc_feature 124..216:
- "misc_feature": Another miscellaneous feature.
- "124..216": Specifies the location from positions 124 to 216 in the sequence.
FT /gene="INS" /note="C-peptide":
- Specifies qualifiers for the C-peptide feature.
- Indicates that the gene associated with this feature is "INS," representing the C-peptide region.
FT misc_feature 223..>285:
- "misc_feature": Another miscellaneous feature.
- "223..>285": Specifies the location, spanning from position 223 to the end of the sequence (>285).
FT /gene="INS" /note="insulin A chain":
- Specifies qualifiers for the miscellaneous feature.
- Indicates that the gene associated with this feature is "INS," representing the insulin A chain.
See Table 1. below for explanation how the locations are specified.
The Sequence
The last item in the feature table is the sequence itself.
SQ Sequence 285 BP; 55 A; 87 C; 93 G; 50 T; 0 other;
ctggggacct gacccagccg cagcctttgt gaaccaacac ctgtgcggct cacacctggt 60
ggaagctctc tacctagtgt gcggggaacg aggcttcttc tacacaccca agacccgccg 120
ggaggcagag gacctgcagg tggggcaggt ggagctgggc gggggccctg gtgcaggcag 180
cctgcagccc ttggccctgg aggggtccct gcagaagcgt ggcattgtgg aacaatgctg 240
taccagcatc tgctccctct accagctgga gaactactgc aacta 285
//
SQ Sequence 285 BP; 55 A; 87 C; 93 G; 50 T; 0 other;
- "Sequence 285 BP": Indicates that the sequence has 285 base pairs.
- "55 A; 87 C; 93 G; 50 T; 0 other": Provides a breakdown of the base composition, indicating the count of each nucleotide (adenine, cytosine, guanine, and thymine) and specifying that there are no other types of bases present. Given the exact base counts, it is easy to compute the GC content of this sequence:
Count(G + C)/Count(G + C + A + T) * 100 = 93+87/93+87+55+50 * 100 = 63.16%
| Location specification | Meaning |
|---|---|
| 42 | A single, specific, known position in the sequence |
| 42..420 | A contiguous span of positions defined by and encompassing the specified boundaries. |
| 42^43 | Indicates a location situated between positions 123 and 124. |
| 42^420 | Indicates a site positioned between two neighboring nucleotides or amino acids, occurring at any location within the range of positions 42 to 420. |
| JF909299:42..420 | Positions 42 through 420, encompassing both ends, within the entry associated with accession number JF909299. |
| complement(42..420) | The complementary sequence corresponds to the segment identified from position 42 to 420 in the sequence record. |
| join(42..72,420..720) | Regions from positions 42 to 72 and 420 to 720 are combined to create a single, uninterrupted sequence. |
| (42.45) | The absence of precise location knowledge. Denotes it to be one of the positions within the range of 42 to 45, inclusively. | <42..420 | A consecutive span of positions with an unknown precise lower boundary; the feature initiates before position 42 but concludes at position 420. |
| >42..420 | The feature initiates somewhere before the 42nd sequenced base and extends up to position 420. | 42..>420 | A continuous span of positions with an unspecified upper boundary; the feature commences at position 42 but concludes somewhere beyond position 420. |
GenBank - Nucleotide Sub-databases
The GenBank database has evolved to encompass additional nucleotide sequences previously housed in separate databases. These include Expressed Sequence Tags (EST), Genome Survey Sequences (GSS), Sequence-Tagged Sites (STS), Reference Sequences (RefSeq), and Reference Gene Sequences (RefSeqGene). The Nucleotide database now facilitates the search for these diverse sequence types.
Expressed Sequence Tags (EST)
Expressed Sequence Tags (EST) transitioned from a standalone database to an integral part of the GenBank database in 2019. Initially, hosting short sequence reads (less than 1000 bp) derived from mRNA (cDNA), dbEST was crucial in capturing a snapshot of genes expressed in specific tissues or under particular conditions. The GenBank-formatted EST record is identified by including "EST" in the KEYWORDS section of the record. The term "EST" is also present on the LOCUS line, signifying the GenBank division.
LOCUS H00099 407 bp mRNA linear EST 26-JAN-2011
DEFINITION 17G01-2 Zinc regulated rat small intestine Rattus norvegicus cDNA
clone 17G01-2 3' similar to human colon EST cm1377 dbj_D25694, mRNA
sequence.
ACCESSION H00099
VERSION H00099.1
DBLINK BioSample: SAMN00154907
KEYWORDS EST.
SOURCE Rattus norvegicus (Norway rat)
...
Figure 2. Part of the header of Genbank flatfile of EST record.
Genome Survey Sequences (GSS)
Genome Survey Sequences (GSS), constituting a GenBank division, serve as a repository for sequences similar to ESTs but originating from genomic DNA rather than cDNA. The repertoire of GSS sequences includes random "single pass read" genome survey sequences, clone end sequences, exon-trapped genomic sequences, Alu PCR sequences, transposon-tagged sequences, and more. The "GSS" keyword is incorporated into the KEYWORDS line of the GenBank-formatted GSS record.
LOCUS JY274301 180 bp mRNA linear GSS 01-JAN-2017
DEFINITION pBCF-6 Maize B-chromosome specific cDNA-AFLP PCR library Zea mays
cDNA clone B4049-180, genomic survey sequence.
ACCESSION JY274301
VERSION JY274301.1
DBLINK BioSample: SAMN01924520
KEYWORDS GSS.
SOURCE Zea mays
...
Figure 3. Part of the header of Genbank flatfile of GSS record.
Sequence-Tagged Sites (STS)
Sequence-Tagged Sites (STS), typically 200 to 500 bp, are generated from polymerase chain reaction (PCR) products. These sequences play a pivotal role in identifying markers such as Simple Sequence Repeats (SSRs), Sequence-Tagged Microsatellite Sites (STMS), Simple Sequence Repeats Polymorphisms (SSRPs), and more. "STS" is in the KEYWORDS line of the Nucleotide STS record and on the LOCUS line.
LOCUS BLYCU 254 bp mRNA linear STS 27-JUL-1995
DEFINITION Hordeum vulgare (clone ABG466) chromosome 4H,5H,6H STS mRNA,
sequence tagged site.
ACCESSION L44026
VERSION L44026.1
KEYWORDS STS; primer; sequence tagged site.
SOURCE Hordeum vulgare subsp. vulgare (domesticated barley)
Figure 4. Part of the header of Genbank flatfile of STS record.
See also GenBank Flafile in the Appendix.
GenBank Divisions
The sequence of records submitted to the GenBank database undergoes systematic categorization into distinct divisions. This categorization is determined by two critical criteria: the taxonomical group of the source organism and the specific sequencing strategy employed to generate the sequencing data. As outlined in Table 2, this comprehensive grouping facilitates a structured organization of genetic information, providing a nuanced understanding of the diverse origins and methodologies associated with the stored genomic data within the GenBank repository.
| Abbrev. | Division Name | Basis of Grouping |
|---|---|---|
| PRI | Primate sequences | Taxonomy |
| ROD | Rodent sequences | Taxonomy |
| MAM | Other mammalian sequences | Taxonomy |
| VRT | Other vertebrate sequences | Taxonomy |
| INV | Invertebrate sequences | Taxonomy |
| PLN | Plant, fungal, and algal sequences | Taxonomy |
| BCT | Bacterial sequences | Taxonomy |
| VRL | Viral sequences | Taxonomy |
| PHG | Bacteriophage sequences | Taxonomy |
| SYN | Synthetic sequences | Taxonomy |
| UNA | Unannotated sequences | Taxonomy |
| EST | EST sequences (expressed sequence tags) | Taxonomy |
| STS | STS sequences (sequence tagged sites) | Taxonomy |
| GSS | GSS sequences (genome survey sequences) | Taxonomy |
| HTG | HTG sequences | Strategy |
| HTC | Unfinished high-throughput cDNA sequencing | Strategy |
| ENV | Environmental sampling sequences | Strategy |
| PAT | Patent sequences | Strategy |
| WGS | Whole genome sequencing projects | Strategy |
References
-
Leinonen R, Akhtar R, Birney E, Bower L, Cerdeno-Tárraga A, Cheng Y, Cleland I, Faruque N, Goodgame N, Gibson R, Hoad G, Jang M, Pakseresht N, Plaister S, Radhakrishnan R, Reddy K, Sobhany S, Ten Hoopen P, Vaughan R, Zalunin V, Cochrane G. The European Nucleotide Archive. Nucleic Acids Res. 2011 Jan;39(Database issue):D28-31. doi: 10.1093/nar/gkq967. PMID: 20972220; PMCID: PMC3013801.
-
Leinonen R, Akhtar R, Birney E, Bower L, Cerdeno-Tárraga A, Cheng Y, Cleland I, Faruque N, Goodgame N, Gibson R, Hoad G, Jang M, Pakseresht N, Plaister S, Radhakrishnan R, Reddy K, Sobhany S, Ten Hoopen P, Vaughan R, Zalunin V, Cochrane G. The European Nucleotide Archive. Nucleic Acids Res. 2011 Jan;39(Database issue):D28-31. doi: 10.1093/nar/gkq967. PMID: 20972220; PMCID: PMC3013801.
-
Yuan D, Ahamed A, Burgin J, Cummins C, Devraj R, Gueye K, Gupta D, Gupta V, Haseeb M, Ihsan M, Ivanov E, Jayathilaka S, Kadhirvelu VB, Kumar M, Lathi A, Leinonen R, McKinnon J, Meszaros L, O'Cathail C, Ouma D, Paupério J, Pesant S, Rahman N, Rinck G, Selvakumar S, Suman S, Sunthornyotin Y, Ventouratou M, Vijayaraja S, Waheed Z, Woollard P, Zyoud A, Burdett T, Cochrane G. The European Nucleotide Archive in 2023. Nucleic Acids Res. 2023 Nov 13: gkad1067. doi: 10.1093/nar/gkad1067. Epub ahead of print.PMID: 37956313.
-
Burgin J, Ahamed A, Cummins C, Devraj R, Gueye K, Gupta D, Gupta V, Haseeb M, Ihsan M, Ivanov E, Jayathilaka S, Balavenkataraman Kadhirvelu V, Kumar M, Lathi A, Leinonen R, Mansurova M, McKinnon J, O'Cathail C, Paupério J, Pesant S, Rahman N, Rinck G, Selvakumar S, Suman S, Vijayaraja S, Waheed Z, Woollard P, Yuan D, Zyoud A, Burdett T, Cochrane G. The European Nucleotide Archive in 2022. Nucleic Acids Res. 2023 Jan 6;51(D1):D121-D125. doi: 10.1093/nar/gkac1051. PMID: 36399492; PMCID: PMC9825583.
-
Cummins C, Ahamed A, Aslam R, Burgin J, Devraj R, Edbali O, Gupta D, Harrison PW, Haseeb M, Holt S, Ibrahim T, Ivanov E, Jayathilaka S, Kadhirvelu V, Kay S, Kumar M, Lathi A, Leinonen R, Madeira F, Madhusoodanan N, Mansurova M, O'Cathail C, Pearce M, Pesant S, Rahman N, Rajan J, Rinck G, Selvakumar S, Sokolov A, Suman S, Thorne R, Totoo P, Vijayaraja S, Waheed Z, Zyoud A, Lopez R, Burdett T, Cochrane G. The European Nucleotide Archive in 2021. Nucleic Acids Res. 2022 Jan 7;50(D1):D106-D110. doi: 10.1093/nar/gkab1051. PMID: 34850158; PMCID: PMC8728206.
-
Tang L. Contamination in sequence databases. Nat Methods. 2020 Jul;17(7):654. doi: 10.1038/s41592-020-0895-8. PMID: 32616930.
-
Sayers EW, Cavanaugh M, Clark K, Ostell J, Pruitt KD, Karsch-Mizrachi I. GenBank. Nucleic Acids Res. 2020 Jan 8;48(D1):D84-D86. doi: 10.1093/nar/gkz956. PMID: 31665464; PMCID: PMC7145611.
-
Sayers EW, Cavanaugh M, Clark K, Ostell J, Pruitt KD, Karsch-Mizrachi I. GenBank. Nucleic Acids Res. 2019 Jan 8;47(D1):D94-D99. doi: 10.1093/nar/gky989. PMID: 30365038; PMCID: PMC6323954.
-
Tanizawa Y, Fujisawa T, Kodama Y, Kosuge T, Mashima J, Tanjo T, Nakamura Y. DNA Data Bank of Japan (DDBJ) update report 2022. Nucleic Acids Res. 2023 Jan 6;51(D1):D101-D105. doi: 10.1093/nar/gkac1083. PMID: 36420889; PMCID: PMC9825463.
-
Sayers EW, Cavanaugh M, Clark K, Ostell J, Pruitt KD, Karsch-Mizrachi I. GenBank. Nucleic Acids Res. 2020 Jan 8;48(D1):D84-D86. doi: 10.1093/nar/gkz956. PMID: 31665464; PMCID: PMC7145611.
-
Okido T, Kodama Y, Mashima J, Kosuge T, Fujisawa T, Ogasawara O. DNA Data Bank of Japan (DDBJ) update report 2021. Nucleic Acids Res. 2022 Jan 7;50(D1):D102-D105. doi: 10.1093/nar/gkab995. PMID: 34751405; PMCID: PMC8689959.
Appendix
GenBank Flafile
Homo sapiens insulin (INS) mRNA, partial cds
GenBank: JF909299.1
FASTA Graphics
Go to:
LOCUS JF909299 285 bp mRNA linear PRI 25-JUL-2016
DEFINITION Homo sapiens insulin (INS) mRNA, partial cds.
ACCESSION JF909299
VERSION JF909299.1
KEYWORDS .
SOURCE Homo sapiens (human)
ORGANISM Homo sapiens
Eukaryota; Metazoa; Chordata; Craniata; Vertebrata; Euteleostomi;
Mammalia; Eutheria; Euarchontoglires; Primates; Haplorrhini;
Catarrhini; Hominidae; Homo.
REFERENCE 1 (bases 1 to 285)
AUTHORS Piri,H., Kazemi,B., Bandehpour,M., Rezaei,M., Goodarzi,M.T.,
Khodadadi,I., Hasanzadeh,T., Sirati Sabet,M., Tasa,D.,
Heidari,H.R., Karimi,J., Yari Khosroshahi,A., Eghdami,A. and
Tavakkoli,A.H.
TITLE Preparation of human insulin gene construct in order to use in
diabetes mellitus gene therapy
JOURNAL Unpublished
REFERENCE 2 (bases 1 to 285)
AUTHORS Piri,H., Kazemi,B., Bandehpour,M., Rezaei,M., Goodarzi,M.T.,
Khodadadi,I., Hasanzadeh,T., Sirati Sabet,M., Tasa,D.,
Heidari,H.R., Karimi,J., Yari Khosroshahi,A., Eghdami,A. and
Tavakkoli,A.H.
TITLE Direct Submission
JOURNAL Submitted (09-APR-2011) Biochemistry, Qazvin University of Medical
Science, Bahonar Blvd., Qazvin 3419759811, Iran
FEATURES Location/Qualifiers
source 1..285
/organism="Homo sapiens"
/mol_type="mRNA"
/db_xref="taxon:9606"
/clone="pBindIns"
/sex="male"
/tissue_type="pancreas"
/country="Iran"
/PCR_primers="fwd_name: INS-fwd, fwd_seq:
cttggaagcttatggccctgtggatgcgc, rev_name: INS-rev, rev_seq:
gcttactcgagctagttgcagtagttctccag"
gene <1..>285
/gene="INS"
CDS <1..>285
/gene="INS"
/note="decreases blood glucose concentration and increases
cell permeability to monosaccharides, amino acids and
fatty acids; accelerates glycolysis, pentose phosphate
cycle, and glycogen synthesis in liver"
/codon_start=2
/product="insulin"
/protein_id="AEG19452.1"
/translation="WGPDPAAAFVNQHLCGSHLVEALYLVCGERGFFYTPKTRREAED
LQVGQVELGGGPGAGSLQPLALEGSLQKRGIVEQCCTSICSLYQLENYCN"
sig_peptide <1..25
/gene="INS"
misc_feature 26..115
/gene="INS"
/note="insulin B chain"
misc_feature 124..216
/gene="INS"
/note="C-peptide"
misc_feature 223..>285
/gene="INS"
/note="insulin A chain"
ORIGIN
1 ctggggacct gacccagccg cagcctttgt gaaccaacac ctgtgcggct cacacctggt
61 ggaagctctc tacctagtgt gcggggaacg aggcttcttc tacacaccca agacccgccg
121 ggaggcagag gacctgcagg tggggcaggt ggagctgggc gggggccctg gtgcaggcag
181 cctgcagccc ttggccctgg aggggtccct gcagaagcgt ggcattgtgg aacaatgctg
241 taccagcatc tgctccctct accagctgga gaactactgc aacta
//
DDBJ Flafile
LOCUS AB000000 450 bp mRNA linear HUM 01-JUN-2009
DEFINITION Homo sapiens GAPD mRNA for glyceraldehyde-3-phosphate
dehydrogenase, partial cds.
ACCESSION AB000000
VERSION AB000000.1
KEYWORDS .
SOURCE Homo sapiens (human)
ORGANISM Homo sapiens
Eukaryota; Metazoa; Chordata; Craniata; Vertebrata; Euteleostomi;
Mammalia; Eutheria; Euarchontoglires; Primates; Haplorrhini;
Catarrhini; Hominidae; Homo.
REFERENCE 1 (bases 1 to 450)
AUTHORS Mishima,H. and Shizuoka,T.
TITLE Direct Submission
JOURNAL Submitted (30-NOV-2008) to the DDBJ/EMBL/GenBank databases.
Contact:Hanako Mishima
National Institute of Genetics, DNA Data Bank of Japan; Yata 1111,
Mishima, Shizuoka 411-8540, Japan
REFERENCE 2
AUTHORS Mishima,H., Shizuoka,T. and Fuji,I.
TITLE Glyceraldehyde-3-phosphate dehydrogenase expressed in human liver
JOURNAL Unpublished (2009)
COMMENT Human cDNA sequencing project.
FEATURES Location/Qualifiers
source 1..450
/chromosome="12"
/clone="GT200015"
/clone_lib="lambda gt11 human liver cDNA (GeneTech.
No.20)"
/db_xref="taxon:9606"
/map="12p13"
/mol_type="mRNA"
/organism="Homo sapiens"
/tissue_type="liver"
CDS 86..>450
/codon_start=1
/gene="GAPD"
/product="glyceraldehyde-3-phosphate dehydrogenase"
/protein_id="BAA12345.1"
/transl_table=1
/translation="MAKIKIGINGFGRIGRLVARVALQSDDVELVAVNDPFITTDYMT
YMFKYDTVHGQWKHHEVKVKDSKTLLFGEKEVTVFGCRNPKEIPWGETSAEFVVEYTG
VFTDKDKAVAQLKGGAKKV"
BASE COUNT 102 a 119 c 131 g 98 t
ORIGIN
1 cccacgcgtc cggtcgcatc gcacttgtag ctctcgaccc ccgcatctca tccctcctct
61 cgcttagttc agatcgaaat cgcaaatggc gaagattaag atcgggatca atgggttcgg
121 gaggatcggg aggctcgtgg ccagggtggc cctgcagagc gacgacgtcg agctcgtcgc
181 cgtcaacgac cccttcatca ccaccgacta catgacatac atgttcaagt atgacactgt
241 gcacggccag tggaagcatc atgaggttaa ggtgaaggac tccaagaccc ttctcttcgg
301 tgagaaggag gtcaccgtgt tcggctgcag gaaccctaag gagatcccat ggggtgagac
361 tagcgctgag tttgttgtgg agtacactgg tgttttcact gacaaggaca aggccgttgc
421 tcaacttaag ggtggtgcta agaaggtctg
//