
Bioinformatics tutorial: Introduction to sequence comparison
Introduction to the following concepts: match, mismatch, substitution, gap, insertion, deletion, indel, global and local alignments.
Apples and oranges.They say that it is not worthwhile to compare apples and oranges, because they are fundamentally different. In any case, it is possible to compare whatever we like. Whether it is worth the effort is another story. Here we are interested in things that we can arrange in sequences; thus we discuss the comparison of sequences.
In general, we can compare two sequences by placing them above each other in rows and comparing them character by character. This way we could align two different audio recordings of a piece of music. There are apps available that can recognize songs by listening to them. In principle, apps accomplish this by alignment of musical notes and rhythms. Programs can detect a similarity between songs the same way, perhaps to identify plagiarism.
Nonetheless, we have a different purpose in bioinformatics, and that is why we will look at the DNA, RNA and protein sequences. Deoxyribonucleic acid (DNA) consists of four bases adenosine, guanine, thymine, and cytosine that are represented by single letters A, G, T, and C respectively. In ribonucleic acid (RNA) thymine (T) is replaced by uracil, represented by the letter U. Protein sequences consist of 20 different amino acids. The single-letter codes for each amino acid are: I, L, V, F, M, C, A, G, P, T, S, Y, W, Q, N, H, E, D, K, R. We will get to that in detail in a later tutorial.
So, why do we compare sequences? There are many reasons, for example, (i) we can identify causes of genetic diseases by comparing sequences from healthy and unhealthy individuals. (ii) A comparison of multiple gene sequences from several species can recognize sequence stretches preserved or similar among species; thus, hinting about the possibility that these conserved regions have a related function in organisms. (iii) Importantly, all sequence database searches involve comparison of sequences to detect a similarity to a search sequence. Note that, the list of reasons is longer than presented here.
Concepts. The following paragraphs introduce the ideas of a match, mismatch, substitution, gap, insertion, deletion, indel, global and local alignments. For the demonstration purposes, we only use DNA sequences, although the concepts also apply to RNA and protein sequences.
Let's take two sequences a) ATGAAGCGTGC, length 11 bases, and b) ATGAAGAGTGCA, length 12 bases. We can make the following alignment as in Figure 1.
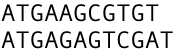
Figure 1. shows pair-wise sequence alignment, but a close examination reveals that we can make the rows to match better. In this alignment, there are only three matching letters on the columns. How can we make additional letters to match?
Gaps. By inserting gaps, the sequences become the same length, and by choosing proper positions, additional letters will align, as shown in Figure 2. In this alignment, nine letters out of the total 13 are aligned and matching in the columns.

The number of matching characters is the same, independent of placement of the gap in position four or five (Figure 3). These two alignments are considered to be separate and different alignments although it is not possible to draw conclusions where to place the gap based on a pair-wise alignment. We may obtain additional information where to position the gap by simultaneously comparing multiple sequences, but this is a topic in multiple sequence alignment tutorial.
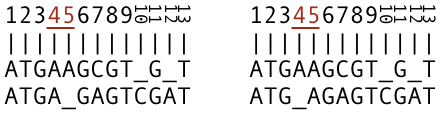
Note the columns four and five, where we placed gaps in the sequence (b). In both cases, we can consider the A to be an insertion into the sequence (a) and a deletion in the same position in the sequence (b). In the columns 10 and 12, we placed gaps into the sequence (a). In this case, sequence (b) has insertions C and A, and sequence (a) deletions in the respective columns. The point is that deletion in one sequence corresponds to insertion in the second sequence and vice versa, but we don't know whether it is an insertion or deletion, because we don't know the exact composition of the parental sequence. For this reason, we call these indels for short. More about this in a later tutorial. Importantly, note that we are not allowed to place gaps in both sequences in the same column. Apart from that, the gaps increase the sequence lengths, possibly to infinity if we like, it is meaningless.

Substitutions. The letters that are aligned and different in the columns are mismatches or substitutions. Our sequences have one substitution C-A in column seven (Figure 4.).
Global sequence alignments. Global sequence alignments are alignments of strings over the entire length of all of them. That is, alignments originate from the start of one sequence, stopping at the end of the other and vice versa. To globally align two divergent sequences or varying lengths often requires insertion of many gaps, which makes the alignments meaningless. Stretching a shorter string may result in dilution of conserved domain alignments, often beyond recognition. Compare the top and bottom alignments in Figure 5, the sequence (b) at the bottom global alignment is the same sequence as in the top only extra characters (to the left and right of the conserved domain) are visible in the bottom alignment. The conserved domains do not align properly. Most often we don't know if sequences match over their whole lengths (Figure 5.). We could try and see, but this is tedious and a waste of time. This where local alignments come to rescue.
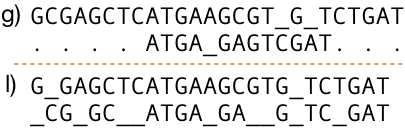
Local sequence alignments. Local sequence alignments consist of alignments of sequences where only a portion of their lengths is aligned (Figure 5. Bottom). Local sequence alignments reduce the number of gaps required in alignments of divergent sequences; thus being able to reveal short conserved or alike regions, that often are undetectable in global sequence alignments. Local sequence alignment algorithms may result in global sequence alignments if the sequences are the same length and very similar. However, global alignment algorithms still have a few uses, such as in the construction of phylogenetic trees for example. That subject you may study in the phylogenetics tree tutorial.
What next?
Related tutorials
Pair-wise sequence alignment
Pair-wise sequence alignment methods
How to select the right substitution matrix?